Chorea Huntington (ICD-10 G10)
Huntington-Krankheit, Veitstanz, Morbus Huntington, danse de St. Guy
- Benannt nach George Huntington, der die Krankheit 1872 im Alter von 22 Jahren beschrieb
- Frühere Beschreibungen durch Elliotson 1832
- 1993 Nachweis des Huntingtin-Gens
Ätiologie
- Autosomal dominante neurodegenerative Erkrankung
- Chromosom 4 (4p16.3, siehe Neurogenetik)
- Degeneration von Globus pallidus, Striatum, Ncl. caudatus, Putamen
- CAG-Repeat (>38) mit Antizipation
- Häufigkeit des CAG-Repeats korreliert invers mit dem Erkrankungsalter
Epidemiologie
- Auftreten in 2-3 Dekade, selten > 60 Jahre
- Prävalenz ca. 5/100000
- Ca. 8000 Erkrankte in Deutschland
Symtome
- Verhaltensstörungen und Psychosen:
- Depression, Ängste, wahnhafte Störungen
- Enthemmung, Reizbarkeit, Delinquenz, Aggressivität
- Bewegungsstörung:
- Chorea (unkontrollierbare, ausfahrende Überbewegungen, Tics, initial an den Extremitäten, Schultern und Gesicht, im Verlauf am ganzen Körper
- Im Schlaf fast vollständiges Sistieren der Bewegungsstörung
- Dementielles Syndrom
- Selten Grand mal
- Westphal-Variante :
- Frühe Manifestation (Kindesalter)
- Rigide Bewegungsstörung
- Rascher Verlauf mit Tod innerhalb weniger Jahre
Besonderheiten bei der klinischen Untersuchung
- Motorische Impersistenz (Zunge kann nicht 10s ruhig herausgestreckt werden)
- Sakkadierende Blickfolge
- Verminderte Konvergenzreaktion
- Gordonsches Kniephänomen (Westphal-Reflex) nach Ausläsen des PSR, langsames Absinken des Unterschenkels
- Erhöhte Ablenkbarkeit: Patient soll ruhigen einen Finger in peripherem Gesichtsfeld fixieren, folgt jedoch unwillkürlich dem anderen bewegten Finger
- Luria-Test: Patient soll fortlaufend nacheinander mit Faust, Handkante und flacher Hand auf den Oberschenkel klopfen
- Pathologischer optokinetischer Nystagmus
- Pseudointendierte Bewegungen - Parakinesie (kaschieren Überbewegung)
- Muskelhypotonie, im Verlauf rigorartige Tonuserhöhung
- Störung der Dysdiadochokinese (Fingerklopfen, alternierende Handbewegungen)
- Ideomotorische Apraxie
- Tics, Überbewegungen
- Sprach- und Schluckstörungen
Therapie - (Detaillierte Informationen erst nach Ärzte-Login)
Um detaillierte Informationen zu den Präparaten zu erhalten loggen Sie sich bitte mit DocCheck in den Ärztebreich ein. (Diese Seite muß dann hierzu neu geladen werden)
- Bewegungsstörung:
- Tiaprid, Haloperidol
- Tetrabenazin
- Tremor: Clonazepam
- Psychische Veränderungen:
- Antidepressiva (SSRI)
- Neuroleptika
- Anxiolytika (Alprazolam, Lorazepam)
Diagnostik
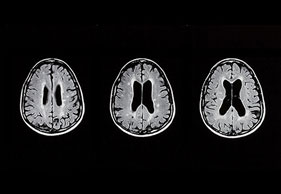
- MRT-Kopf (Erweiterung der Vorderhörner, Atrophie Straitum)
- Vorstellung in humangenetischer Sprechstunde
- Genetische Diagnostik (siehe Neurogenetik)
- SSEP (Med./Tib.): Geminderte Amplituden
- Blinkreflex: Verzögerte R2-Latenz
- Testpsychologie: WCST, Turm von Hanoi u.a.
- Ev. ENG
- Ev. SPECT: Striataler Hypometabolismus
Verlauf
- Initial häufig psychische Veränderungen mit leichten Bewegungsstörungen
- Im Verlauf Zunahme der Bewegungsstörung und Hinzutreten der Demenz
- Progredienter Verlauf mit letalem Ausgang
- Erkrankungsdauer 10-25 Jahre
Differentialdiagnose
- Spinocerebelläre Ataxie (Typ 17), Choreo-Akanthozytose, Dystonien, Dementielle Erkrankungen
